
Grok your data
The objective of bioSyntax is to bring you closer to your data, giving you an intuitive & empathetic understanding of biology. To appreciate all that bioSyntax has to offer read this short manual (~10 minutes) and go explore.
- Getting Started
- Reading Data
- Supported File Formats
- Support
- Collaborating on bioSyntax
- See also: bioSyntax Manuscript
Getting Started
See: Installing bioSyntax
bioSyntax integreates seamlessly with vim (Linux / Mac / Win), sublime (Linux / Mac / Win), gedit (Linux / Win), & less (Linux / Mac). After installing bioSyntax files will automatically detected by file-extension.
Reading large-data
For very large data sets, it’s often slow to open them in a text editor. It’s best to use the command-line program less which will read your file from a data-stream.
Read your large-data set with less directly
# If your file is uncompressed, it can be read directly.
# less will recognize the file extension (.XYZ)
cd ~/myData/
less dbSNP107_common.vcf
less hg19.fa
Streaming your data directly into less with pipes |
# If your file is compressed, you can 'pipe' the data
# using the "|" operator from decompression, directly into
# less. You must prefix the file extension you want
# as file formats are not recognized within streams.
cd ~/myCompressedData/
samtools view -h NA12878_hg38.bam | sam-less
gzip -dc dbSNP107_rare.vcf.gz | vcf-less
gzip -dc hg38.fa.gz | fa-less
Bypassing bioSyntax (data in plain-text)
For vim
Type :syntax off in vim
For less
# You may want to view your data without syntax highlighting
# such as where a file is improperly formatted or very large
# files where syntax highlighting may be slow (i.e. VCF files
# with hundreds of columns).
# 1. Pipe your data through cat
cat snp_1000genomes.vcf | less -
# 2. Within less, switch to a visual editor
less snp_1000genomes.vcf
# press 'CTRL-C' to stop process
# press 'v' to switch to visual editor
Reading Data
Nucleotides
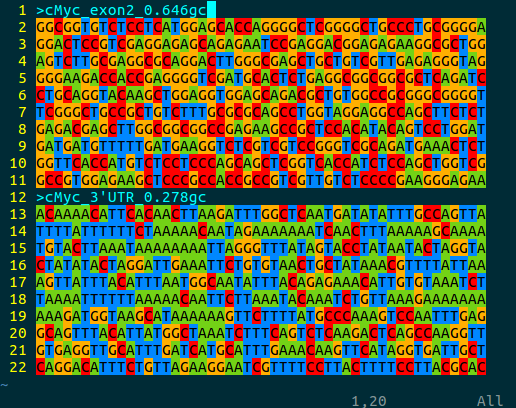
bioSyntax implements a novel, full IUPAC Nucleotide Code coloring. Ambiguous bases are represented by an ~additive color-mixing of the parent bases. For example, Thymine (blue) + Cytosine (red) are both pYrimidines (magenta).
An intuitive feature of the bioSyntax color scheme is that the ‘GC-content’ of a sequence can be quickly approximated by how warm (high GC, red-orange) or cool (low GC, blue-green) a sequence looks.
vim myc_gcContent.fa
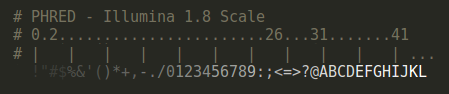
PHRED Scores
When available, bioSyntax will highlight PHRED quality scores in a step-gradient of blacks (PHRED = 0-10) to whites (PHRED = 40+).
CIGAR Strings
In .sam files the Query:Reference alignment is summarized efficiently but illegibly as a CIGAR String. With a little bit of highlighting these become much easier to read.

Amino Acid Color Schemes
You can choose from several color-schemes for amino-acid fasta files. The Fasta Clustal (Default) syntax colors amino acids based on their physiochemical properties, so does Fasta Hydrophobicity, or you may prefer better discrimination of each amino acids with Fasta Zappo or Fasta Taylor.
Supported File Formats
File format and software compatibility matrix for bioSyntax.
| status | |
|---|---|
| X | Syntax Complete |
| o | In Development |
| - | Unavailable |
Core bioSyntax
| File Format | Description | sublime | vim | gedit | less |
|---|---|---|---|---|---|
| .fasta | Generic nt/aa sequence | X | X | X | X |
| .fastq | Fasta + PHRED quality | X | X | X | X |
| .clustal | Multiple Sequence Alignment | X | X | X | X |
| .bed | Genomic Ranges | X | X | X | X |
| .gtf | Genomic Annotation | X | X | X | X |
| .pdb | Protein Structure | X | X | X | X |
| .vcf | Variant Call Format | X | X | X | X |
| .sam | NGS Sequence Data | X | X | X | X |
Auxillary Syntaxes
| File Format | Description | sublime | vim | gedit | less |
|---|---|---|---|---|---|
| .fasta | fasta alternative AA colors | ||||
| - | Clustal | X | - | X | - |
| - | Taylor | X | - | X | - |
| - | Zappo | X | - | X | - |
| - | Hydrophobicity | X | - | X | - |
| .fai | Fasta Index (faidx) | X | X | X | X |
| .flagstat | samtools flag summary | X | X | X | X |
| .cwl | Common Workflow Language | X | X | X | - |
| .wig | Wiggle data | - | - | X | - |
| .nexus | Phylogenetics data | - | X | - | - |
| .pml | Pymol Script Language | X | X | - | - |
See Also: Alternative/User Syntax Definitions
These syntaxes are not part of the unified bioSyntax suite but often serve specialized functions.
Science Syntaxes
| File Format | Description | sublime | vim | gedit | less |
|---|---|---|---|---|---|
| .gaussian | Gaussian File (chemistry) | - | X | - | - |
If you’d like to add support for another file-format; check the development page to get started.
Support
Report a bug / Ask a question
The fastest way to get an answer is to:
1) Search / Open an issue on the bioSyntax Repo.
Please Include:
- A detailed and descriptive title.
- Enough information about what did for someone else to replicate the problem.
- Information about the operating system / software you’re using (
uname -a) - If it’s a syntax highlighting issue: a screenshot of the error and a small bit of the input file you used.
2) If you really don’t want to make a (fake) github account. Email [email protected] and we’ll open the issue, but it will be slower.
Uninstallation Instructions
Collaborating on bioSyntax
bioSyntax is a community-oriented project for scientific syntax highlighting. We encourage you to change and customize it to suit your needs.
Check out the Development page to create syntax-highlighting for custom file-formats and for other ways to help out.